
Hashimoto’s Thyroiditis as Thyroid-Cell Toxicity: An ROS-Centered View
Hashimoto's is typically viewed as an autoimmune attack on the thyroid. Here I present a new theory of Hashimoto's, viewing it as toxic overload of oxidative stress that damages thyroid cells.

Welcome to my blog! If you’re new here, please keep an open mind. Our goal is an ambitious one: we are trying to completely reinterpret the nature of chronic disease. The deeper we dig into this topic, the more it seems that toxins, and the oxidative stress they create, are the ultimate drivers of disease.
We saw this for cancer, where we viewed cancer as a symptom of toxic overload. We also saw this for diabetes, where we interpreted Type 1 and Type 2 diabetes as the destruction of the pancreatic beta cells caused by acute and chronic toxic overload, respectively.
In the case of diabetes, we found that the pancreatic beta cells exhibited a striking vulnerability to Reactive Oxygen Species (ROS). In this blog post, I will turn to another specialized endocrine gland that shows striking parallels: the thyroid. The thyroid also is surprisingly vulnerable to the oxidative stress from toxic overload.

Interestingly, both the pancreas and the thyroid have associated “autoimmune diseases”, which correspond to Type 1 diabetes (T1D) and Hashimoto’s thyroiditis, respectively. I have already argued that T1D should be viewed as a disease of toxicity, rather than as an autoimmune condition. In this blog, I will make the same argument for Hashimoto’s. This raises deep questions about autoimmune diseases more broadly.
Hashimoto’s is the most common cause of hypothyroidism in the modern world. So I believe it’s an exciting breakthrough to have a new theory for Hashimoto’s, which I call Thyroid-Cell Toxicity Theory (TCTT). This represents another step towards a unified theory of chronic disease that’s centered around toxicity and ROS.
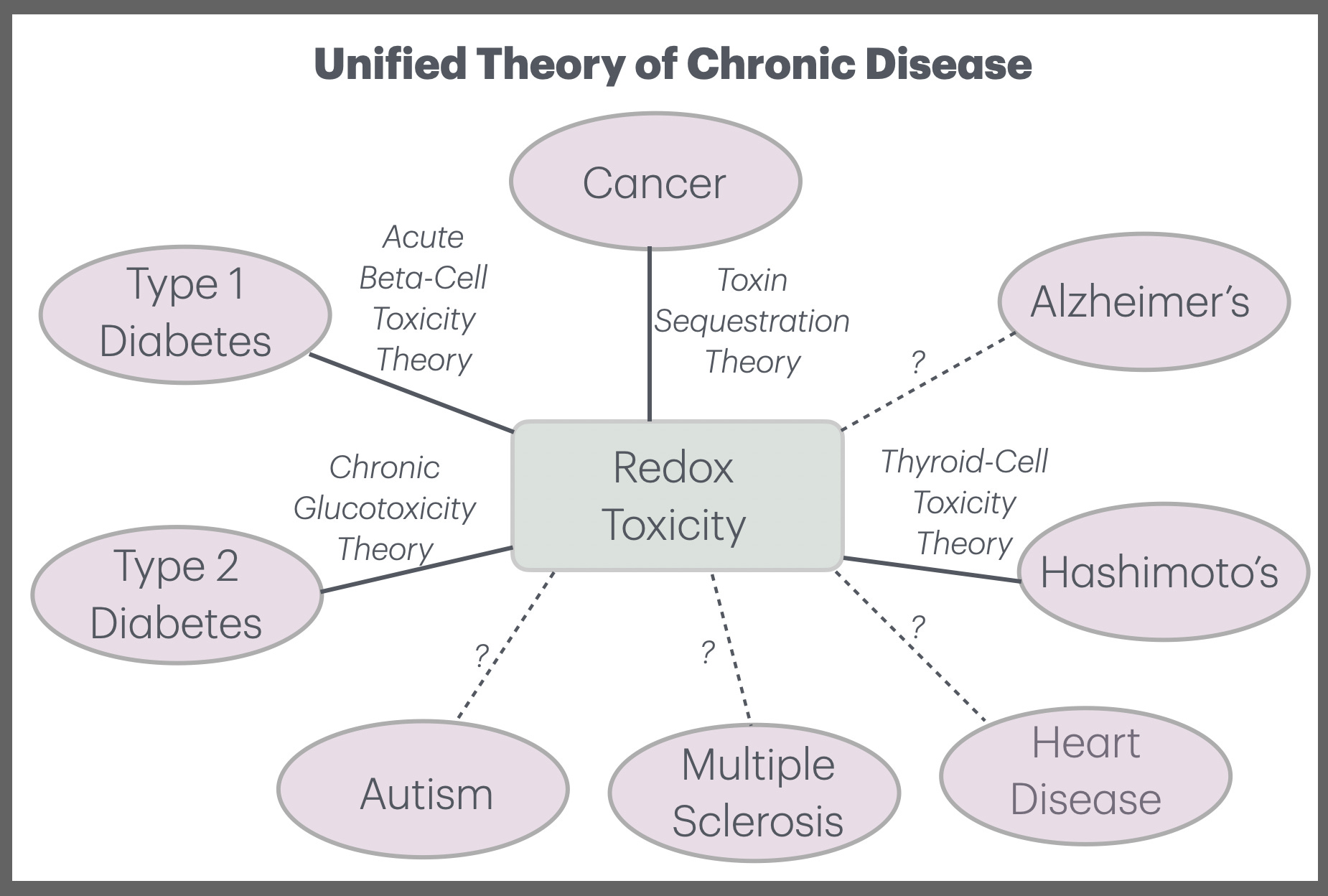
What Is Hashimoto’s Thyroiditis?
Hashimoto’s is a chronic inflammatory condition characterized by progressive damage to the thyroid gland, resulting in reduced production of thyroid hormones.
Clinically, it often presents with symptoms such as fatigue, weight gain, cold intolerance, dry skin, hair loss, constipation, brain fog, and depression. Many patients also develop a goiter (enlarged thyroid). Laboratory findings typically include elevated thyroid peroxidase (TPO) and thyroglobulin (TG) antibodies, rising TSH levels, and eventually low free T4 as the disease progresses.
While it can occur at any age, Hashimoto’s is far more common in women than in men (roughly 5 to 10 times higher incidence) and often appears between the ages of 30 and 60. Once significant thyroid tissue is lost, patients usually require lifelong thyroid hormone replacement therapy.
In conventional medicine, Hashimoto’s is classified as an autoimmune disease. However, as we will explore in this post, a growing body of evidence suggests that oxidative stress and ROS overload may play a more fundamental role — with the autoimmune response frequently acting as a secondary cleanup mechanism rather than the primary cause.
The Mainstream View – and Its Limitations
Mainstream medicine describes Hashimoto’s as an autoimmune disease. In this framework, the immune system mistakenly identifies components of the thyroid gland as foreign and mounts a sustained attack, leading to chronic inflammation, progressive destruction of thyroid tissue, and eventual hypothyroidism.
Genetic predisposition (particularly certain HLA genes) is considered the primary risk factor, while environmental influences such as viral infections, stress, or iodine intake are thought to act as triggers that initiate or accelerate the autoimmune process in susceptible individuals. The strong female predominance of the disease is viewed as the result of estrogen-driven immune hyperactivity combined with X-chromosome genetic effects.
This model accounts for many clinical observations, including the high levels of TPO and thyroglobulin antibodies, and the lymphocytic infiltration seen on biopsy. Treatment typically focuses on thyroid hormone replacement and, in some cases, monitoring or modulating the immune response.
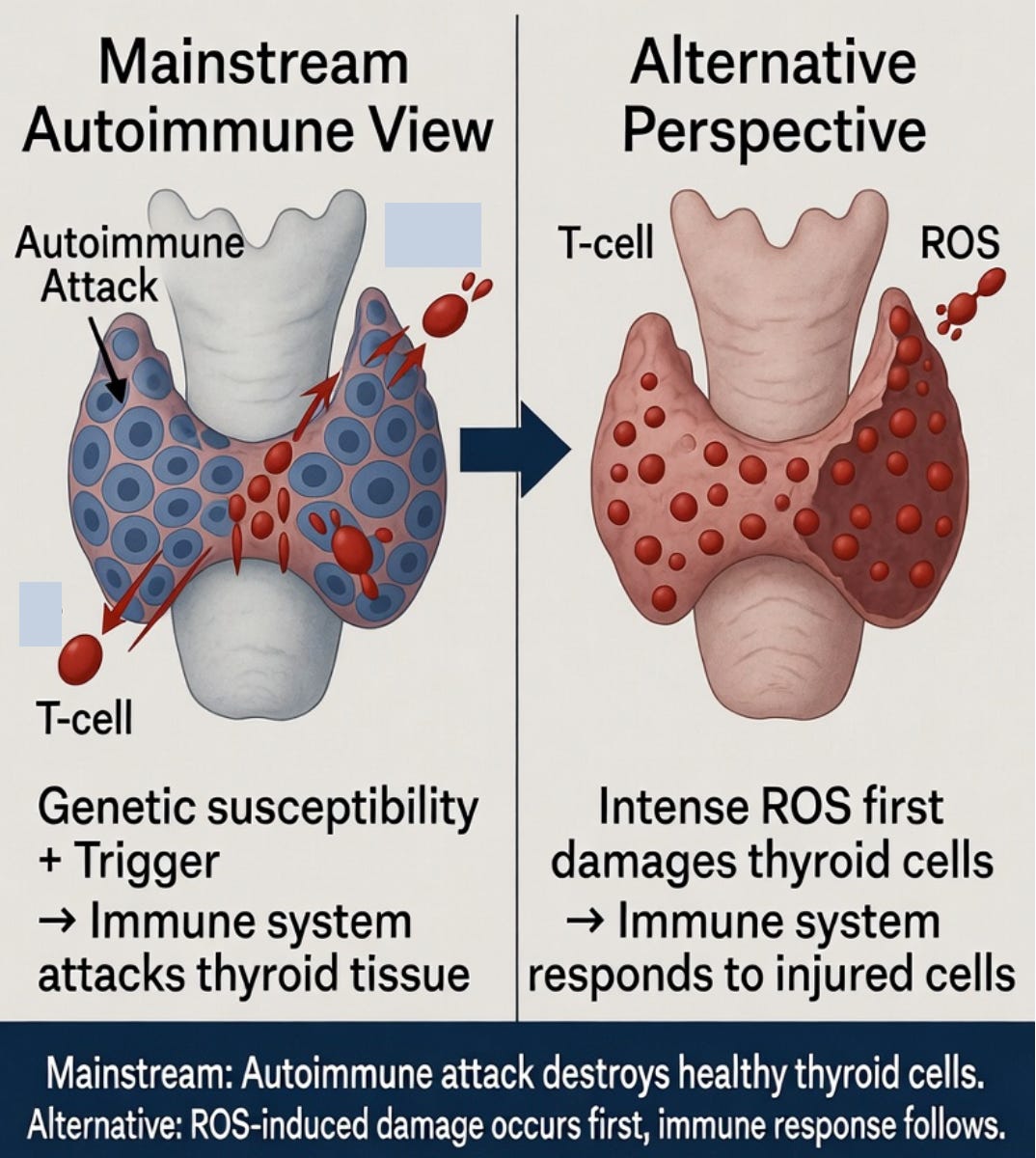
However, this autoimmune-centric view leaves several important questions unanswered. Why does the thyroid gland appear so selectively vulnerable compared to other tissues? Why do patients often show elevated markers of oxidative stress even in early stages of the disease? And why is there such wide variation in disease progression and severity, even among individuals with similar genetic backgrounds?
These gaps suggest that something more fundamental may be occurring upstream of the immune response — namely, chronic or acute oxidative stress that damages thyroid follicular cells first, setting the stage for the secondary immune reaction that follows. This motivates a new theory: Thyroid-Cell Toxicity Theory.
Thyroid-Cell Toxicity Theory (TCTT)
Let’s now state the core principles of TCTT:
Thyroid follicular cells are uniquely vulnerable to ROS.
Hashimoto’s begins with oxidative damage to thyroid cells.
Multiple toxins contribute to the total redox burden.
The autoimmune response is often secondary.
Hypothyroidism results from cumulative redox damage.
In what follows, I will dive into each of these principles.
Why Thyroid Follicular Cells Are Uniquely Vulnerable to ROS
Thyroid follicular cells have one of the most redox-challenging jobs in the body: they deliberately generate large amounts of hydrogen peroxide (H₂O₂) every day as an essential step in thyroid hormone synthesis.
This constant production of H₂O₂, mediated by DUOX enzymes and thyroid peroxidase, creates an inherently high-oxidative environment inside the gland. While this process is necessary for making T4 and T3, it also puts the cells at constant risk of oxidative spillover. To manage this, the thyroid relies heavily on selenium-dependent antioxidant enzymes such as glutathione peroxidase. When these defenses are insufficient, excess H₂O₂ can easily convert into more damaging reactive oxygen species (ROS).
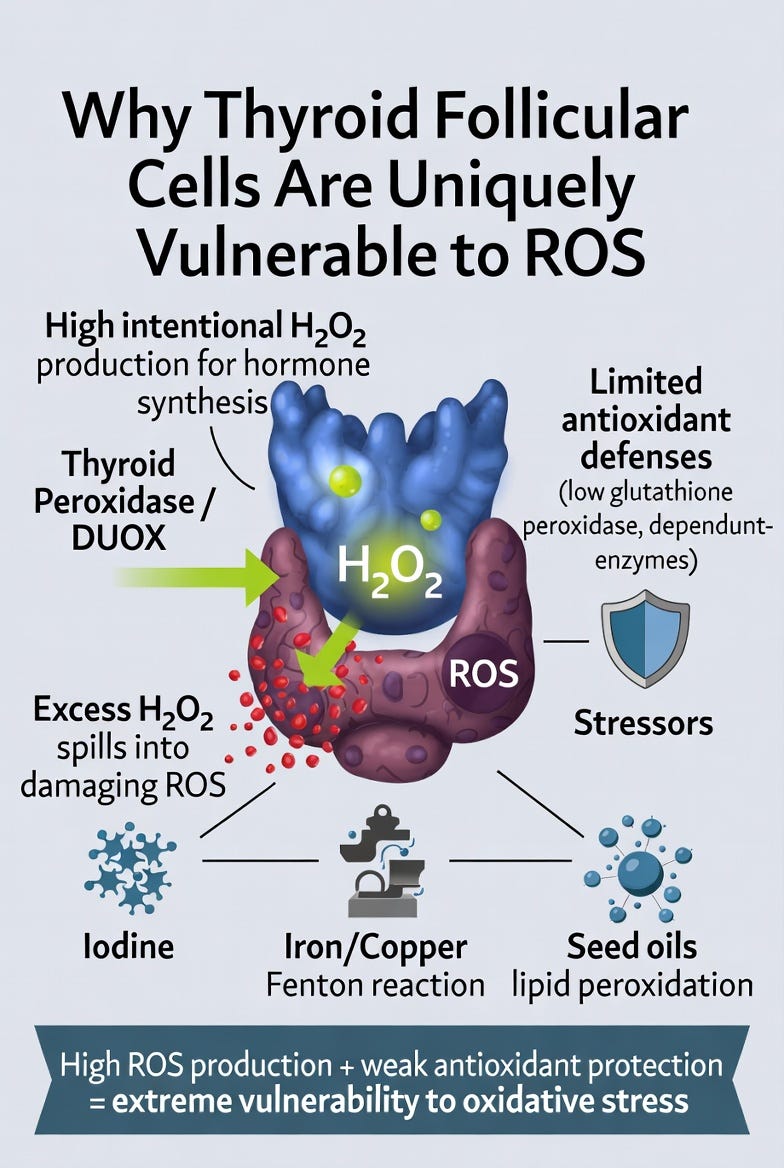
Compared to most other tissues, thyroid follicular cells combine three dangerous factors:
High intentional ROS production
Relatively modest baseline antioxidant capacity
Constant exposure to environmental stressors that further amplify oxidative stress
This unique vulnerability explains why the thyroid is so sensitive to redox imbalance. Just as beta cells are the weak link in glucose regulation, thyroid follicular cells are the weak link in thyroid hormone regulation. When ROS production exceeds the gland’s ability to neutralize it, cellular damage accumulates rapidly.
Hashimoto’s as Thyroid-Cell Toxicity: The Core Mechanism
In the ROS-centered view, Hashimoto’s thyroiditis is best understood as thyroid-cell toxicity — the progressive or acute failure of thyroid follicular cells under excessive oxidative stress.
The thyroid gland naturally operates in a high-oxidative environment. Every day, follicular cells generate large amounts of hydrogen peroxide (H₂O₂) to produce thyroid hormones. When additional ROS-generating stressors — such as excess iodine, iron or copper overload, seed oils, mycotoxins, or halide competition — push the total redox burden too high, the gland’s limited antioxidant defenses become overwhelmed.
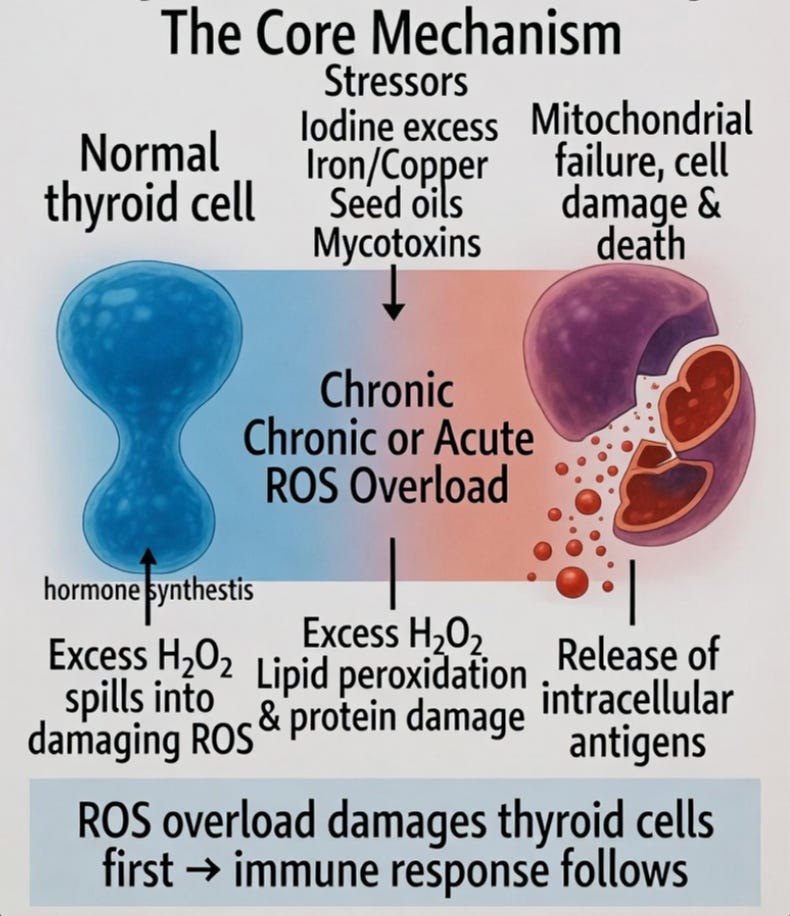
Excess H₂O₂ spills over into damaging reactive oxygen species, causing mitochondrial dysfunction, lipid peroxidation of cell membranes, and direct injury to thyroid cells. Over time, critical proteins and structures are damaged, leading to cell dedifferentiation, apoptosis, or necrosis. As thyroid cells are injured and die, they release intracellular antigens (including thyroid peroxidase and thyroglobulin), which the immune system then recognizes and targets.
What appears on biopsy and blood tests as an “autoimmune attack” is often a secondary cleanup response to already damaged tissue rather than an unprovoked assault on healthy cells. In this framework, the primary event is oxidative injury to the thyroid’s hormone-producing units, followed by the immune system’s reaction to the resulting cellular debris.
This mechanism parallels the Acute Beta-Cell Toxicity Theory in diabetes: the specialized endocrine cells fail first under ROS overload, and the immune response follows.
Common Triggers of ROS Overload in the Thyroid
Because the thyroid naturally operates in a high-hydrogen-peroxide environment, it is particularly susceptible to additional sources of oxidative stress. When these stressors push the total redox burden beyond the gland’s limited antioxidant capacity, thyroid follicular cells can suffer progressive or rapid damage.
Several key triggers stand out:
Iodine excess significantly increases H₂O₂ production during hormone synthesis. In susceptible individuals, this can lead to oxidative spillover and direct cellular injury.
Iron and copper overload are especially problematic. In the thyroid’s high-H₂O₂ setting, these metals readily catalyze the Fenton reaction, generating highly reactive hydroxyl radicals that damage mitochondria, membranes, and proteins.
Excess estrogen (including from hormonal birth control or endogenous estrogen dominance) upregulates ROS-generating enzymes such as DUOX and NOX4, increasing hydrogen peroxide production and oxidative stress in the thyroid.
Seed oils rich in linoleic acid contribute through lipid peroxidation, which amplifies oxidative stress within thyroid tissue.
Mycotoxins (such as aflatoxins and ochratoxins) and certain heavy metals (mercury, cadmium, arsenic) can directly impair mitochondrial function and deplete glutathione.
Other halides like bromide and fluoride may act as competitive inhibitors of iodine uptake, leading to inefficient hormone synthesis and compensatory increases in ROS production.
Radiation, from sources such as dental X-rays or medical imaging, delivers an acute burst of ROS through water radiolysis. Because the thyroid is located in the neck, it often receives scatter radiation during dental procedures.
Goitrogens found in cruciferous vegetables (like broccoli, kale, and cabbage) can interfere with iodine uptake and thyroid peroxidase activity. In theory, very high intakes of raw cruciferous vegetables could increase oxidative inefficiency and compensatory H₂O₂ production, adding to the thyroid’s redox burden.
Oxalate crystals are frequently deposited in thyroid tissue and are found at higher rates in Hashimoto’s patients. They cause local inflammation and add to the gland’s baseline oxidative burden.
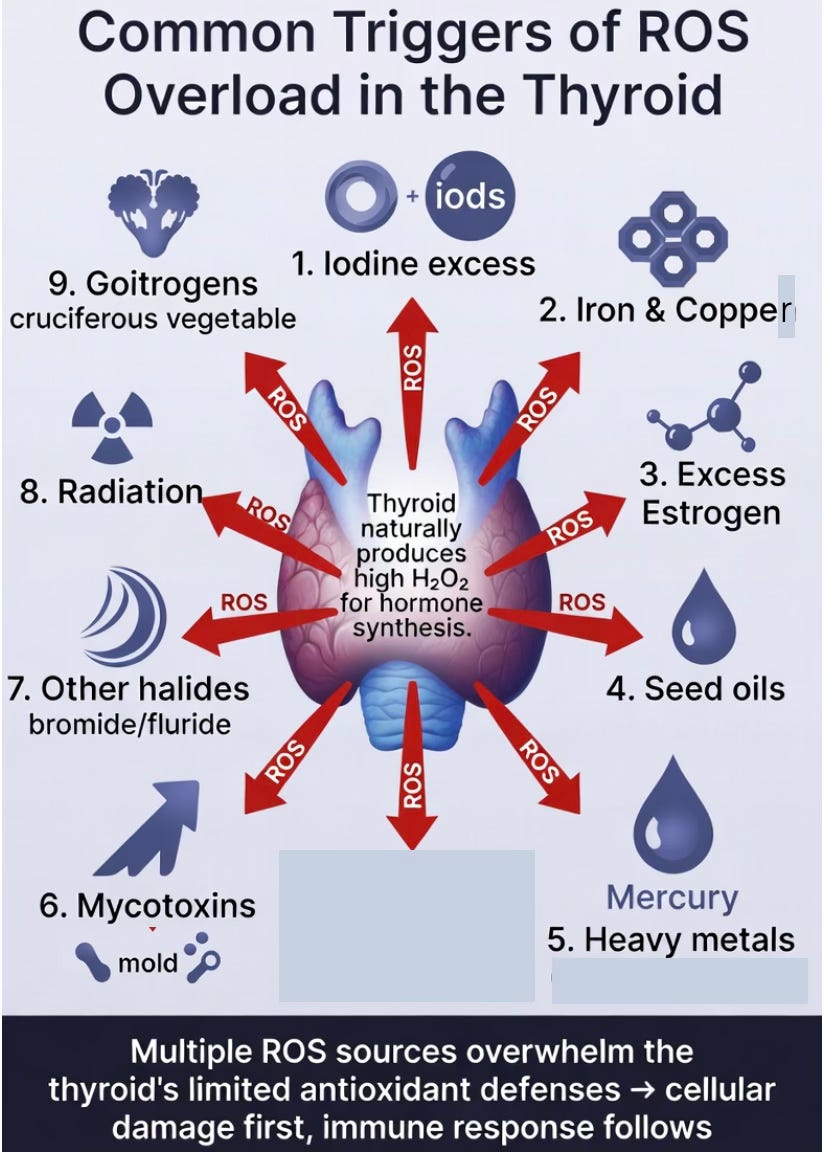
In the thyroid-cell toxicity view, these triggers rarely act alone. They often combine to create cumulative redox pressure. When this pressure exceeds the gland’s defenses, follicular cell damage occurs first — setting the stage for the secondary immune response that characterizes Hashimoto’s
Protective Mechanisms and Their Limits
The thyroid gland is not defenseless. It has built-in protective mechanisms designed to handle its naturally high oxidative environment. The most important defense is the selenium-dependent antioxidant system — particularly glutathione peroxidase and thioredoxin reductase — which neutralizes excess hydrogen peroxide and prevents it from spilling into damaging ROS. Adequate selenium status is therefore critical for protecting thyroid follicular cells. However, these protective systems have clear limits. When the total ROS burden increases, the gland’s antioxidant capacity can be overwhelmed. Chronic low selenium intake, common in many modern diets, further weakens this defense. Once these protective mechanisms are exceeded, oxidative damage accumulates in thyroid cells.
The Autoimmune Response Is Often Secondary
In TCTT, the immune attack seen in Hashimoto’s is not the primary cause of the disease. Instead, it is a secondary response. Excessive ROS first damages thyroid follicular cells. Once these cells are injured or dying, the immune system is activated to clear the damaged tissue and cellular debris. What appears as an “autoimmune” attack is, in many cases, the body’s natural cleanup process following oxidative injury. This reframing helps explain why immune-suppressing treatments often provide only partial or temporary relief — they address the cleanup crew while leaving the underlying redox overload untouched. Previously, we saw the same pattern appear in Type 1 diabetes, where beta-cell destruction precedes the autoimmune response.
Hypothyroidism results from cumulative redox damage
In TCTT, hypothyroidism in Hashimoto’s is not caused by the immune system destroying the thyroid. Instead, it is the predictable outcome of cumulative redox damage. When the thyroid’s natural high-H₂O₂ environment is repeatedly stressed by toxins like excess iodine, iron and copper, excess estrogen, seed oils, heavy metals, mycotoxins, radiation, and goitrogens, follicular cells gradually suffer oxidative injury. Over time, enough cells are damaged or lost that the gland can no longer produce adequate thyroid hormones. The autoimmune response seen on blood tests and biopsy is largely secondary — the immune system arriving to clear away the injured and dying cells. Thus, Hashimoto’s hypothyroidism represents the final stage of a long, quiet process of redox overload overwhelming the thyroid’s limited defenses. The immune attack is not the root cause; it is the cleanup after sustained toxic damage.
Why Hashimoto’s Is Far More Common in Women
Hashimoto’s shows one of the strongest sex biases in medicine, affecting women 5 to 10 times more frequently than men. This disparity is not adequately explained by immune system differences alone.
Women experience significantly higher lifetime exposure to estrogen, which has a direct and potent effect on thyroid redox balance. Estrogen upregulates key enzymes (DUOX and NOX4) that increase hydrogen peroxide production inside thyroid follicular cells. At the same time, estrogen strongly induces the liver to produce more ceruloplasmin, the main copper-carrying protein in blood. As a result, women typically maintain higher serum copper levels than men. In the thyroid’s already high-hydrogen-peroxide environment, this elevated copper catalyzes the Fenton reaction, converting H₂O₂ into highly damaging hydroxyl radicals.
This estrogen-copper synergy creates a higher baseline redox burden in the female thyroid. The effect becomes especially pronounced during periods of hormonal fluctuation — puberty, pregnancy, postpartum, and perimenopause — when estrogen and copper levels rise and fall together. Many women first develop Hashimoto’s during or shortly after these transitions. Modern hormonal birth control and copper IUDs may further amplify this burden by increasing estrogen exposure and adding direct copper, respectively.
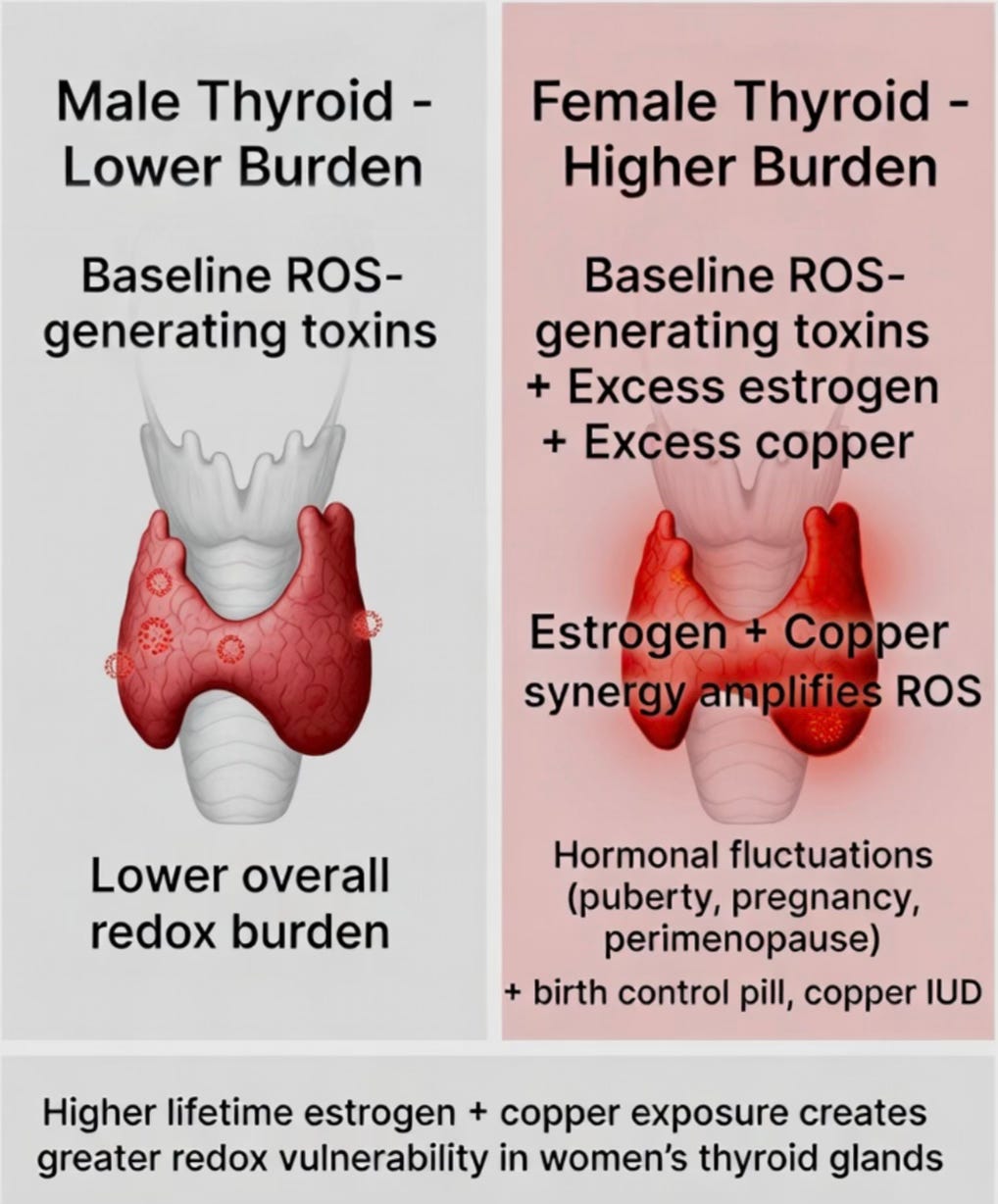
This does not mean women have universally higher rates of all ROS-related diseases. Rather, the thyroid is uniquely vulnerable because it is one of the few organs that intentionally runs a high-H₂O₂ system for normal function. When estrogen and copper amplify that system, the gland becomes a target for redox damage. In TCTT, the higher prevalence in women reflects this specific redox vulnerability rather than a generally “more autoimmune” immune system.
The Iodine controversy
Iodine remains the most debated topic in thyroid health. Excess iodine clearly increases hydrogen peroxide (H₂O₂) production in thyroid follicular cells — a necessary step for thyroid hormone synthesis. In susceptible individuals, this can contribute to oxidative stress and trigger or worsen Hashimoto’s.
However, iodine is rarely the sole culprit. In TCTT, excess iodine acts primarily as additional fuel for the fire rather than the fire itself. The real danger arises when elevated H₂O₂ meets other redox toxins — particularly iron and copper. These metals catalyze the Fenton reaction, converting relatively mild hydrogen peroxide into highly destructive hydroxyl radicals (•OH) that damage cellular components. Without these amplifying factors, many people can tolerate higher iodine intake reasonably well, especially when supported by adequate selenium (which helps clear H₂O₂ efficiently).
Notably, Dr. David Brownstein advocates high-dose iodine protocols for Hashimoto’s. While these protocols have produced anecdotal benefits for some patients, they carry significant risk for many others. By dramatically increasing H₂O₂ production in an already stressed gland, high-dose iodine can accelerate oxidative damage when iron, copper, or other redox toxins are present.

This explains why some individuals appear to handle high-dose iodine without issues, while others experience rapid worsening. The outcome depends heavily on the total redox burden — not just the amount of iodine.
On a personal note, I briefly tried Brownstein’s high-dose iodine protocol in 2024, and it gave me major side effects. I had never had thyroid issues in my life, but once on his protocol, I experienced hypothyroid symptoms. Needless to say, I admit my mistake, and I do not recommend this protocol to anyone.
Reinterpreting Autoimmune Diseases
Having reinterpreted both Type 1 diabetes and Hashimoto’s as conditions driven by ROS-mediated cellular toxicity rather than an autoimmune attack, an important question arises: Could many other autoimmune diseases follow a similar pattern? In both cases, the sequence appears consistent — oxidative damage to a specific cell type occurs first, followed by a secondary immune response that clears the damaged cells. The immune system is responding to injury, not initiating it.
If this pattern holds more broadly, many conditions currently labeled “autoimmune” (such as rheumatoid arthritis, multiple sclerosis, or lupus) may ultimately reflect the body’s attempt to manage chronic or acute redox stress in vulnerable tissues. The immune response may be downstream of an underlying toxin-driven oxidative burden rather than the root cause.

This perspective does not diminish the suffering caused by these diseases, but it shifts the focus toward identifying and reducing the upstream sources of redox overload. It suggests a more unified and hopeful framework: many autoimmune conditions may be protective responses in the face of modern toxic burdens. Further exploration of this idea could open new avenues for prevention and treatment.
Implications for treatment
If Hashimoto’s is driven by ROS overload rather than an autoimmune process, then treatment should focus on lowering the total redox burden on the thyroid gland and supporting its protective mechanisms. This means reducing exposure to excess iodine, iron, copper, heavy metals, seed oils, mycotoxins, oxalates, bromide / fluoride, and radiation. Ensuring adequate selenium status is especially important. For many patients, these steps can reduce oxidative damage to thyroid cells, potentially slow disease progression, and in some cases improve residual thyroid function.
Mainstream medicine primarily relies on immune-suppressing strategies (such as steroids or other immunomodulators) and lifelong thyroid hormone replacement. TCTT suggests a hopeful shift: Hashimoto’s may not be an inevitable lifelong autoimmune disease, but rather a preventable or modifiable state of redox overload in a highly sensitive gland. Early intervention focused on lowering the oxidative load may help preserve thyroid function and improve long-term outcomes.
Conclusion
In summary, TCTT offers a more unified and hopeful framework for Hashimoto’s. It suggests that many “autoimmune” conditions may actually begin with oxidative toxicity rather than immune dysfunction. By identifying and reducing the upstream sources of redox stress, we may be able to prevent or slow the progression of Hashimoto’s and similar diseases. True healing likely lies not in stronger immune suppression, but in lightening the toxic load on the thyroid — and on the body as a whole.
Thanks for reading! Let me know your thoughts!
Hi Patrick thank you for your good work! I too am enjoying the benifits of Carnivore. My mind is much sharper than before. Do you feel this carnivore lifestyle has helped you think through this amazing information? Im thinking so.
Botany driven carnavore Ken
Would you say this is also the same concept for Graves’ disease